

Abstract
Background: Langerhans cell sarcoma (LCS) is a rare malignant neoplasm of Langerhans cells characterized by multi-organ involvement and an aggressive clinical course.Given the rarity of the disease, the true epidemiological data on LCS are lacking, and most of our knowledge is obtained from institutional case series. In this study, we utilized two national databases- the Surveillance, Epidemiology, and End Results (SEER) Program database (https://seer.cancer.gov/) and National Cancer Data Base (NCDB) to study the incidence, clinical presentation, and outcomes of LCS.
Methods: We calculated LCS incidence (cases/10, 000, 000 individuals) from SEER database using SEER*Stat (v 8.3.4; https://seer.cancer.gov/seerstat/) statistical software. SEER is a population-based registry program of the United States National Cancer Institute that covers approximately 28% of the population. We identified LCS cases that were confirmed histologically using International Classification of Diseases for Oncology edition 3 (ICD-0-3) histology code 9756/3 from SEER 18 (2000-2014) registry. As the disease was mandatory reportable only after 2001, we included cases that were diagnosed after 2001 in the analysis. Demographic patterns, clinical presentation, and overall survival (OS) were calculated utilizing the patient level data from the NCDB Participant User File. NCDB is a joint program by the American College of Surgeons and American Cancer Society that represents approximately 70% of newly diagnosed cancer cases in the nation. All patients aged > 18 years with a diagnosis LCS (ICD-0-3 Code 9756/3) from 2004 to 2015 were included. To ensure accuracy of follow-up, we excluded the patients that were not treated at the same facility of diagnosis from survival analysis.
Results: A total of 25 cases of LCS were reported to SEER between 2000 and 2014. The overall incidence of the disease was 0.2 per 10,000,000 [95% CI:0.1-0.3] and was similar among males and females (p=0.33). The incidence of the disease did not differ significantly among different races (whites, blacks and other races) (p=0.56).
Between 2004 and 2015, 52 patients with newly diagnosed LCS were identified from NCDB and 60% were males. The median age of diagnosis was 62 (range, 19-90 years). The most common sites of the presentation were connective tissue (29%; n=15), reticuloendothelial system (RES) and hematopoietic system (25%; n=13), skin (19%, n=10), gastrointestinal (10%, n=5), bones and joints (8%, n=4), brain (2%, n=1) and breast (2%, n=1). Site specific details were missing in 27% (n=14) of cases. Out of the 52 patients, twenty patients (39%) received chemotherapy as first-line therapy, while 24 (46%) received surgery and 15 (29%) received radiation therapy.
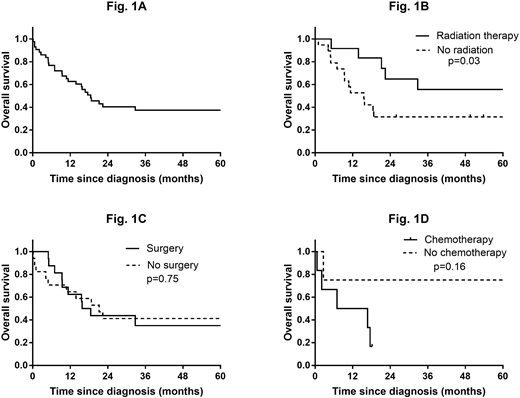
After a median follow up of 18 (range, 1-144) months, 27 patients (52%) had died. The one- year survival rate was 62%, and the median OS was 19 months (Fig. 1A). We also calculated the 1-year disease-specific survival and OS using SEER database, and was 74% and 63%, respectively. After censoring the patients with hematopoietic and RES involvement, the patients who were managed with radiation therapy (median dose 3960 cGy, range 750-6400) had a better OS as compared to those who received no radiation therapy (p=0.03, Fig. 1B). On the contrary, after excluding the patients with hematopoietic and RES as primary site, no significant difference in OS was noted between the patients who were managed with and without surgery (17 vs 21 months; p = 0.75; Fig. 1C). In addition, post-surgical radiation or chemotherapy had no significant benefit in median OS (p=0.25). Among patients with hematopoietic and RES involvement, there was no association between OS and receipt of chemotherapy (p = 0.16; Fig. 1D); however, the number of patients who received chemotherapy was small (n=6).
Conclusion: This dual-national registry study shows that LCS is extremely rare, and has a poor prognosis. Although the data regarding optimal management of LCS are limited, our study demonstrates that radiation therapy may offer survival advantage in patients without hematopoietic and RES involvement.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal